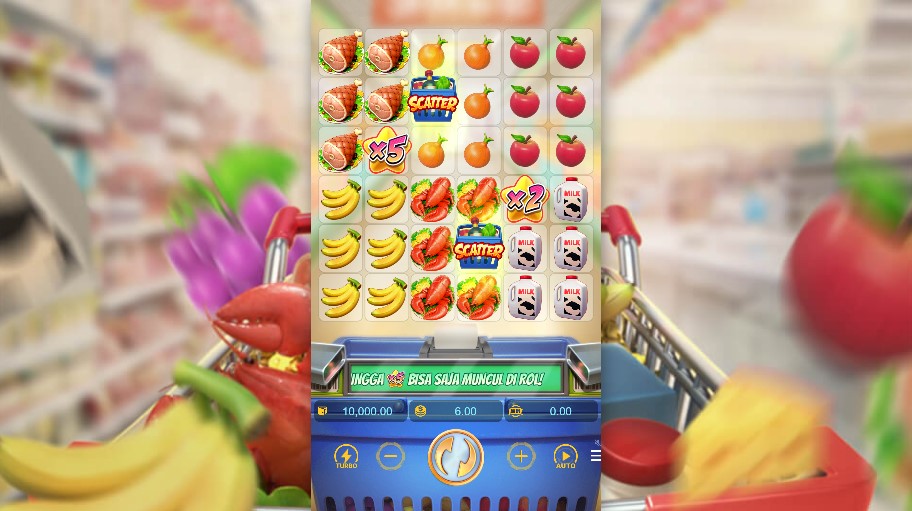
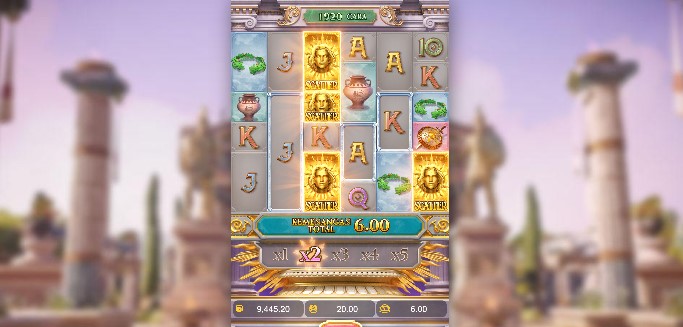
Situs Slot Demo Gacor Gampang Maxwin Fortune Rabbit
Situs Slot Demo Gacor Gampang Maxwin Fortune Rabbit  Link Slot Gacor Terpercaya Menang Maxwin Emotiwins
Link Slot Gacor Terpercaya Menang Maxwin Emotiwins  Situs Slot Judi Online Jackpot Terbesar Flying High
Situs Slot Judi Online Jackpot Terbesar Flying High  Situs Judi Slot Gacor Mudah Menang Caishen’s Cash
Situs Judi Slot Gacor Mudah Menang Caishen’s Cash  Link Situs Slot Terpercaya Jackpot Terbesar Cheeky Emperor
Link Situs Slot Terpercaya Jackpot Terbesar Cheeky Emperor 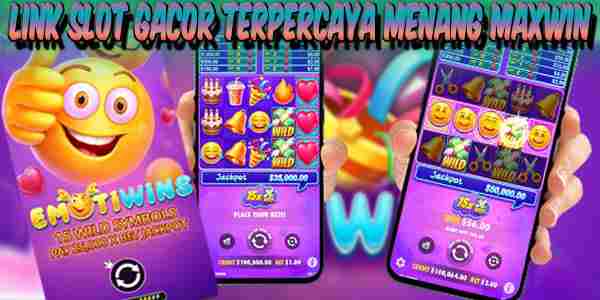









Situs Slot Demo Gacor Gampang Maxwin Fortune Rabbit
Situs Judi Slot Demo Gacor Resmi Terpercaya Gampang Maxwin Fortune Rabbit
Untuk para member setia kami, sebaik lebih dahulu mencoba situs link slot demo gacor resmi terpercaya. Game slot online menawarkan keseruan yang tidak kalah dengan permainan uang sungguhan. Dengan berlatih secara rutin, keterampilan Anda akan semakin berkembang.
Link judi slot demo gacor ini, memberi kesempatan untuk mencoba game slot online terbaru dari pengembang terkemuka. Ini adalah cara pertama terbaik untuk mengetahui judi slot terbaik yang memiliki peluang menang terbesar.
Bocoran Link Live RTP Slot Online Tertinggi Hari Ini

Game online Fortune Rabbit memiliki RTP tinggi, menjadikan bettor meraih jackpot terbesar dengan mudah. RTP adalah faktor penting dalam menemukan game slot gacor gampang menang. Dapatkan informasi terbaru tentang game judi slot demo terbaik mudah menang.
Dalam artikel ini, kami akan memberikan bocoran RTP slot tertinggi malam ini yang wajib Anda coba. Oleh karena itu, memilih permainan slot online jackpot terbesar untuk meningkatkan peluang menang Anda.
Untuk informasi lebih lengkap, silakan kunjungi situs slot gacor terpercaya mudah menang kami.
Maksimalkan Fitur Game Judi Slot Online24Jam Terbaik
Nantinya para bettor dapat menikmati fitur game judi slot terbaik dengan peluang menang besar. Fitur-fitur seperti multiplier, free spin, dan tumbler menjadi daya tarik utama dalam permainan slot online. Dengan adanya fitur-fitur ini, pemain dapat meningkatkan peluang mereka untuk memenangkan hadiah besar.
1️⃣ Fitur Tumble Slot Demo Anti Lag
Tumble game slot memungkinkan simbol-simbol pemenang untuk menghilang dan memberi ruang bagi simbol baru untuk jatuh ke posisi tersebut. Setiap kali simbol baru jatuh dan membentuk kombinasi yang menang, pemain bisa memperoleh kemenangan berulang dalam satu putaran. Fitur tumble memberikan peluang lebih besar untuk meraih kemenangan tambahan tanpa perlu memasang taruhan lagi.
2️⃣ Fitur Multiplier Aplikasi Slot 777
Multiplier adalah fitur yang dapat meningkatkan kemenangan Anda dengan mengalikannya menggunakan angka tertentu. Misalnya, jika Anda menang dengan multiplier 2x, kemenangan Anda akan digandakan menjadi dua kali lipat. Faktanya multiplier ini sangat dicari pemain karena memberi kesempatan untuk meraih kemenangan lebih besar dari taruhan awal.
3️⃣ Fitur Free Spin Aplikasi Slot 888
Free spin adalah salah satu fitur yang paling dicari oleh pemain judi online. Fitur ini memungkinkan Anda untuk memutar gulungan slot tanpa mengeluarkan uang tambahan. Free spin terpicu setelah memenuhi syarat tertentu dalam permainan, seperti mencocokkan simbol scatter.
Bergabunglah dengan pemain yang sudah berpengalaman dalam bermain judi slot online. Selanjutnya mulailah petualangan bettor dalam link slot demo gacor terbaik kami.